CAR-T細(xì)胞療法已經(jīng)在血液惡性腫瘤中顯示出令人印象深刻的療效。去年,,美國FDA批準(zhǔn)了兩種靶向CD19的CAR-T細(xì)胞療法,,諾華的tisagenlecleucel(Kymriah™)用于白血病(2017年8月)和淋巴瘤(2018年5月),以及Kite Pharma的axicabtagene ciloleucel(Yescarta™)用于淋巴瘤(2017年10月),。
這也推動了在實(shí)體瘤中誘導(dǎo)相似功效的CAR的發(fā)展,。然而,該過程在實(shí)現(xiàn)足夠的功效之前,,還面臨著多個(gè)必須解決的挑戰(zhàn):
在實(shí)體瘤中CAR-T細(xì)胞治療的眾多挑戰(zhàn)中,一個(gè)主要障礙是缺乏真正的腫瘤特異性靶抗原,,這迫使細(xì)胞免疫學(xué)家靶向在腫瘤上過表達(dá)的腫瘤相關(guān)抗原(tumor-associated antigens, TAAs),,但由于TAA也在正常組織和器官表達(dá),具有安全風(fēng)險(xiǎn),。
此外,,實(shí)體瘤的腫瘤微環(huán)境(tumor microenvironment, TME)特別是免疫抑制劑,阻止有效的抗腫瘤免疫應(yīng)答,。免疫抑制性TME含有多種成分,,包括物理屏障,如致密的細(xì)胞外基質(zhì),;功能失調(diào)的上皮細(xì)胞,;代謝檢查點(diǎn),如缺氧,;免疫屏障,,如免疫抑制細(xì)胞因子/分子和免疫抑制性免疫細(xì)胞。
近日,,CAR-T領(lǐng)域“大牛”賓夕法尼亞大學(xué)的Carl June教授在Frontiers in Immunology上發(fā)表了綜述,,針對目前CAR-T細(xì)胞治療實(shí)體瘤的嚴(yán)峻形勢,指出:為了有效地靶向?qū)嶓w瘤,,必須同時(shí)解決影響功效和毒性的多種因素,,深入了解CAR-T細(xì)胞生物學(xué)和影響CAR-T細(xì)胞療法治療窗的多種因素。
Carl June教授(圖片來源:Philadelphia Magazine)
在這篇綜述中,,Carl June等人總結(jié)了CAR抗原識別機(jī)制的新發(fā)現(xiàn),,還討論了調(diào)整和擴(kuò)展治療窗,使CAR-T細(xì)胞有效且安全地靶向?qū)嶓w瘤的合理策略,。
或許,,能給正在實(shí)體瘤CAR-T細(xì)胞療法中掙扎的你一些有用的信息,。
CAR-T細(xì)胞生物學(xué)基礎(chǔ)知識
雖然已經(jīng)深入研究了T細(xì)胞通過TCR與靶標(biāo)相互作用的基本機(jī)制,但CAR與靶標(biāo)相互作用的機(jī)制尚不清楚,。CAR由TCR復(fù)合物和抗體的組合部分組成,,因此討論CAR與內(nèi)源性、未修飾的TCR-T細(xì)胞的相似性,,以及定義CAR的明顯差異能更好地理解CAR-T細(xì)胞生物學(xué),。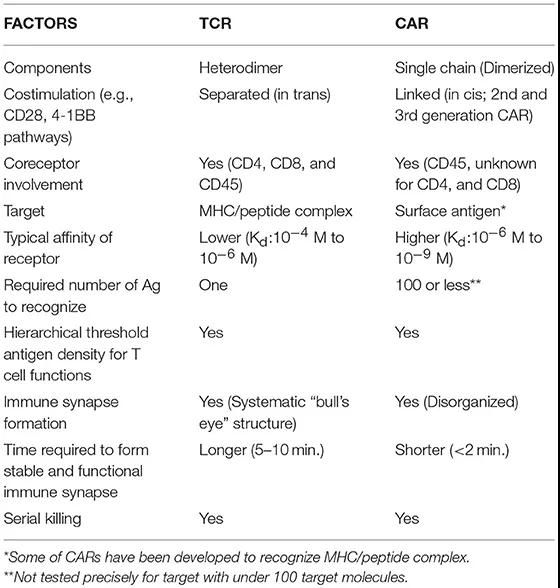
CAR-T和TCR-T細(xì)胞生物學(xué)因素的比較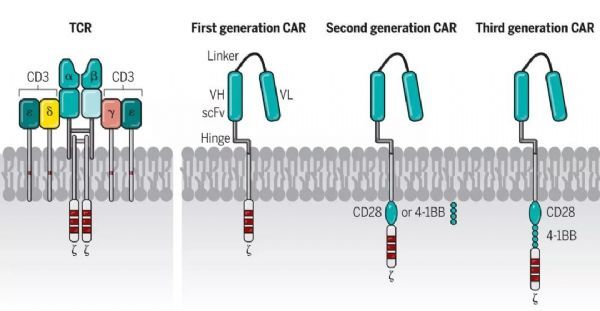
TCR與CAR的結(jié)構(gòu)對比(圖片來源:Science)
>>>>TCR
由TCRα和TCRβ亞基組成的異二聚體。每個(gè)亞基含有可變區(qū)結(jié)構(gòu)域(V)和恒定區(qū)結(jié)構(gòu)域(C),,其后是跨膜區(qū),。每個(gè)V結(jié)構(gòu)域含有三個(gè)互補(bǔ)決定區(qū)(CDR),其與主要組織相容性復(fù)合物(MHC)上呈遞的肽相互作用,。
TCR本身不具有信號結(jié)構(gòu)域,,需要由CD3復(fù)合物啟動細(xì)胞內(nèi)信號傳導(dǎo)。CD3復(fù)合物由三個(gè)二聚體組成,,分別是CD3ζε,、CD3δε異二聚體和CD3ζζ同二聚體。
>>>>CAR
合成的嵌合蛋白,,被引入T細(xì)胞以重定向抗原特異性并增強(qiáng)細(xì)胞功能,。通常地,CAR由來自mAb的單鏈可變片段(scFv),、細(xì)胞外間隔區(qū)(稱為鉸鏈),、跨膜結(jié)構(gòu)域、CD3ζ信號傳導(dǎo)結(jié)構(gòu)域和通常一或兩個(gè)(第二代或第三代)共刺激結(jié)構(gòu)域組成,。
非典型構(gòu)建的CAR利用受體配體或肽作為細(xì)胞外抗原識別結(jié)構(gòu)域,,例如zetakine CAR:白細(xì)胞介素-13受體α2(IL13Rα2)zetakine CAR。
CAR賦予T細(xì)胞以不依賴于MHC的方式通過scFv(抗體識別)直接結(jié)合表面抗原的益處,。CAR可以通過CD3ζ和共刺激結(jié)構(gòu)域同時(shí)向T細(xì)胞傳遞信號,,這可以誘導(dǎo)T細(xì)胞的化學(xué)計(jì)算的和潛在理想的活化。
CAR如何觸發(fā)免疫突觸形成和傳遞信號,?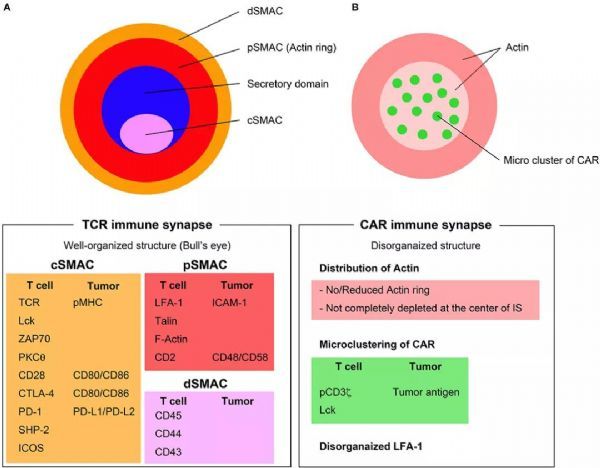
通過TCR和CAR形成免疫突觸
>>>>
TCR IS
T細(xì)胞活化是通過TCR與MHC-肽復(fù)合物(稱為IS)的高度有組織的和動態(tài)的相互作用介導(dǎo)的,。成熟的IS是基于TCR信號的聚集體,誘導(dǎo)T細(xì)胞應(yīng)答,。
IS由三個(gè)同心的聚集分子環(huán)組成(圖A):
IS的內(nèi)環(huán)被稱為中心超分子激活簇(cSMAC),,其中發(fā)生TCR信號傳導(dǎo)。cSMAC含有大多數(shù)TCR-MHC-肽復(fù)合物,、CD28,、PKC-θ和Lck;
外周SMAC(pSMAC)含有參與細(xì)胞粘附的蛋白質(zhì),,例如整聯(lián)蛋白LFA-1,、細(xì)胞骨架連接子talin和ICAM1,;
大分子,如CD43和CD45,,被排除在pSMAC之外,,構(gòu)成遠(yuǎn)端SMAC(dSMAC);
裂解顆粒的分泌發(fā)生在細(xì)胞毒性T淋巴細(xì)胞(CTL)和靶細(xì)胞之間的IS變體(即分泌性突觸)內(nèi),。分泌性突觸在cSMAC中具有兩個(gè)獨(dú)立且不同的結(jié)構(gòu)域:(1)包含信號蛋白的信號傳導(dǎo)結(jié)構(gòu)域,;(2)與細(xì)胞因子、穿孔素和顆粒酶的胞吐作用有關(guān)的分泌結(jié)構(gòu)域,。Stinchcombe等人證明,,由Lck信號控制的瞬時(shí)極化和中心體對質(zhì)膜的對接,在指導(dǎo)這種分泌的機(jī)制中具有重要作用,。
此外,,抑制和共刺激分子,如PD-1,、CTLA-4和ICOS也在IS區(qū)域聚集,,并在調(diào)節(jié)T細(xì)胞活化中發(fā)揮關(guān)鍵作用。
>>>>
CAR IS
CAR下游的細(xì)胞內(nèi)信號傳導(dǎo)和CAR形成IS的機(jī)制尚未得到廣泛研究,。
已經(jīng)證明,在CD19特異性CAR-T細(xì)胞和靶細(xì)胞之間,,ZAP70向IS募集以及CD45被排除在IS之外,,類似于TCR活化。TCR的下游信號分子,,如CD3ζ,、LAT、Lck和ZAP70在CD19特異性CAR-T細(xì)胞被自體CD19+ B細(xì)胞激活后磷酸化,。與第二代CAR-T細(xì)胞相比,,第三代CAR-T細(xì)胞在TCR的下游信號分子上具有顯著更高的磷酸化狀態(tài)。
CAR IS(圖B)與TCR IS的結(jié)構(gòu)不同,。CAR IS沒有呈現(xiàn)出系統(tǒng)性的靶眼結(jié)構(gòu),,而這是TCR IS的一個(gè)顯著特征。CAR IS肌動蛋白(Actin)環(huán)的組織性很差,,并且肌動蛋白在CAR IS的中心可能不會*消失,。LFA-1紊亂,CAR腫瘤抗原復(fù)合物在CAR IS的微團(tuán)簇形成隨機(jī)分布,。
TCR IS需要5-10分鐘來形成靶眼結(jié)構(gòu),,而CAR IS可能不需要形成這些穩(wěn)定的結(jié)構(gòu),因?yàn)镃AR IS的無組織多焦點(diǎn)模式足以快速誘導(dǎo)顯著的近端信號傳導(dǎo),,其發(fā)生在短時(shí)間內(nèi)(<2分鐘),。
IS生物學(xué)的另一個(gè)重要部分是將細(xì)胞毒性顆粒(包括穿孔素和顆粒酶)遞送至由微管組織中心(MTOC)介導(dǎo)的IS,。CAR的近端信號傳導(dǎo)的快速但短暫的持續(xù)時(shí)間也誘導(dǎo)MTOC快速遷移至IS并加速顆粒的遞送。
雖然CAR IS的機(jī)制已逐漸顯現(xiàn),,但尚不清楚CAR IS結(jié)構(gòu)的差異是否與CAR-T細(xì)胞的功效相關(guān),。
可溶形式CAR的配體,如CD30,、間皮素和CEA,,存在于單體形式不能觸發(fā)CAR信令,因?yàn)樗鼈儾粫餋AR二聚化,。然而,,CAR-T細(xì)胞可能潛在地識別可以以寡聚形式存在的可溶性配體,例如TGF-β,,甚至無需細(xì)胞-細(xì)胞相互作用,。
CAR-T細(xì)胞識別的目標(biāo)密度閾值是多少?
為了解決CAR激活閾值的問題,,Watanabe等人研究了激活CD20特異性CAR-T細(xì)胞(CD28共刺激結(jié)構(gòu)域)所需的CD20密度,,其中每個(gè)靶細(xì)胞表達(dá)約200-250,000個(gè)CD20分子。在細(xì)胞內(nèi)表達(dá)低密度CD20(約200個(gè)分子/細(xì)胞)的靶細(xì)胞即可被CAR-T細(xì)胞誘導(dǎo)裂解,。該數(shù)據(jù)與之前的報(bào)道一致,,即CAR靶向小鼠OTS8的腫瘤特異性糖表位,其可以裂解具有相似低密度(~200分子/細(xì)胞)靶抗原的靶細(xì)胞,。
Watanabe等人還證明誘導(dǎo)T細(xì)胞增殖和細(xì)胞因子產(chǎn)生所需的靶抗原密度要高于誘導(dǎo)CAR介導(dǎo)的裂解所需的靶抗原密度,。
總之,CAR-T細(xì)胞可識別具有相當(dāng)?shù)退降陌锌乖陌屑?xì)胞,,并且它們具有針對細(xì)胞裂解,、增殖和個(gè)體細(xì)胞因子產(chǎn)生的分級T細(xì)胞信號傳導(dǎo)閾值。CAR構(gòu)建體的差異(例如,,scFv,,鉸鏈,共刺激結(jié)構(gòu)域的親和力)或CAR表達(dá)密度,,可以用于更地控制CAR-T細(xì)胞活化,。
CAR-T細(xì)胞是否可以作為連環(huán)殺手?
內(nèi)源性T細(xì)胞和NK細(xì)胞可以連續(xù)裂解多個(gè)靶細(xì)胞(串聯(lián)殺傷),,這很可能*腫瘤所必需的,。然而,直到近,,CAR-T細(xì)胞介導(dǎo)連續(xù)殺傷的能力和靶細(xì)胞裂解的動力學(xué)才得到充分證實(shí),。
Davenport等人使用一種新的轉(zhuǎn)基因小鼠模型測試了通過內(nèi)源性TCR或異位表達(dá)的CAR激活的細(xì)胞毒性T淋巴細(xì)胞(CTL)的功能。他們使用定時(shí)顯微錄影術(shù)(time-lapse video microscopy)清楚地證明CAR-T細(xì)胞是連續(xù)殺手。大約22%的CAR-T細(xì)胞依次向兩個(gè)或三個(gè)腫瘤細(xì)胞遞送致死命中,,并且通過CAR連續(xù)殺死的頻率與通過TCR的頻率相當(dāng),。
腫瘤細(xì)胞裂解的動力學(xué)分析表明,在前20小時(shí)內(nèi),,TCR和CAR介導(dǎo)等效的裂解動力學(xué),。但與20小時(shí)后TCR介導(dǎo)的裂解相比,CAR介導(dǎo)的細(xì)胞裂解動力學(xué)減慢,。這種差異可以通過在抗原識別刺激后的CAR下調(diào)來解釋,,這可以通過基于TCR的CAR表達(dá)來改善。
CAR親和力如何影響T細(xì)胞功能,?
T細(xì)胞活化受TCR和MHC-肽復(fù)合物之間相互作用的調(diào)節(jié),,并且影響活化敏感性的主要因素是靶抗原密度和TCR親和力。在微環(huán)境中腫瘤的主要免疫抑制機(jī)制是抗原識別失敗,,原因是低親和力的TCR和癌癥相關(guān)的肽-MHC復(fù)合物的相互作用,。
TCR對自身衍生肽(如腫瘤抗原)的親和力低于TCR對病原體衍生抗原的親和力。因此,,通常更難以分離對TAA具有足夠敏感性的T細(xì)胞,,這是過繼性細(xì)胞療法(ACT)的個(gè)障礙。
另一方面,,TCR的高親和力伴隨著自身免疫應(yīng)答,,當(dāng)患者接受ACT治療時(shí),有時(shí)會導(dǎo)致嚴(yán)重的不良事件,。已經(jīng)報(bào)道,,ACT利用超生理的、高親和力的TCR不能提高療效,,反而MHC-肽復(fù)合物的數(shù)量少能夠?qū)崿F(xiàn)高TCR占用,原因是單個(gè)復(fù)合體可以串聯(lián)接合并觸發(fā)數(shù)百個(gè)TCR,。
總之,,這表明一種模型,其中TCR的理想親和力應(yīng)提供足夠長時(shí)間的相互作用來實(shí)現(xiàn)近端信號的傳導(dǎo),,但也應(yīng)適當(dāng)?shù)亟档陀H和力,,以分離并允許盡可能多的TCR遇到MHC-肽復(fù)合物。
scFv親和力對CAR-T細(xì)胞功能反應(yīng)的影響仍未*了解,。通常,,與天然TCR親和力相比,用scFv構(gòu)建的CAR具有更高的親和力,。由于大多數(shù)TAA在腫瘤上高表達(dá),,在正常組織中低表達(dá),增加CAR的親和力將可能導(dǎo)致on-target/off-tumor效應(yīng)引起的嚴(yán)重不利影響的風(fēng)險(xiǎn),因此必須考慮刺激閾值以獲得CAR-T細(xì)胞活化的*特異性,。
與天然T細(xì)胞類似,,CAR-T細(xì)胞也可以以連續(xù)方式殺死多個(gè)靶細(xì)胞。當(dāng)通過CAR刺激而不是通過TCR刺激腫瘤細(xì)胞時(shí),,可以更快地消除腫瘤細(xì)胞,,因?yàn)镃AR可以比TCR更快地從垂死的腫瘤細(xì)胞中解離出來。因此,,增加CAR-T細(xì)胞的親和力可能減少/防止連續(xù)殺傷,,促進(jìn)T細(xì)胞衰竭,并減少中樞記憶和效應(yīng)表型T細(xì)胞的產(chǎn)生和持久性,,或通過激活誘導(dǎo)細(xì)胞死亡來增加T細(xì)胞的損失等一系列不利影響,。
實(shí)體瘤靶抗原的篩選
ACT的關(guān)鍵是選擇靶抗原,以提供足夠的功效并使毒性小化,。一些針對腫瘤特異性抗原的CAR已在臨床前開發(fā)當(dāng)中,,包括針對異常糖基化癌基因的CAR,如MUC1的Tn糖型,,以及腫瘤特異性活化形式的整合素,,以及臨床上,如CARs靶向膠質(zhì)母細(xì)胞瘤中的腫瘤特異性轉(zhuǎn)錄變體EGFRvIII,。
在沒有更多腫瘤特異性靶標(biāo)的情況下,,CAR-T細(xì)胞療法很可能選擇繼續(xù)靶向在正常組織中也表達(dá)的實(shí)體瘤TAA。實(shí)際上,,大多數(shù)正在進(jìn)行的針對實(shí)體瘤的CAR-T細(xì)胞療法的臨床試驗(yàn)都針對這樣的TAA,。
了解正常組織是否表達(dá)抗原及其表達(dá)水平以預(yù)測潛在毒性至關(guān)重要。
基于基因表達(dá)(RNA測序或微陣列)或免疫組織化學(xué)(IHC),,可獲得正常組織上抗原表達(dá)的幾個(gè)公共數(shù)據(jù)庫,。但是,這些技術(shù)包含限制和陷阱,。對于基因表達(dá)分析,,非常罕見但關(guān)鍵細(xì)胞表達(dá)的抗原可能被低估。此外,,可能無法區(qū)分表達(dá)的基因是來自組織還是來自浸潤細(xì)胞,。此外,假陽性和假陰性是尚未解決的問題,,IHC對低表達(dá)抗原的敏感性可能不足以選擇實(shí)體瘤的CAR靶標(biāo),。
新技術(shù),如單細(xì)胞RNA測序,,可以提供更準(zhǔn)確的表達(dá)譜,,使研究人員能夠更好地預(yù)測新型CAR-T細(xì)胞的功效和毒性,。
擴(kuò)大CAR-T細(xì)胞療法治療窗的策略
“治療窗”是藥物毒理學(xué)的術(shù)語,定義為功效和毒性之間的一系列劑量,,實(shí)現(xiàn)高治療益處并且不會導(dǎo)致不可接受的毒性,。盡管工程化細(xì)胞與傳統(tǒng)藥物的藥代動力學(xué)大不相同,但將治療窗的概念應(yīng)用于ACT領(lǐng)域?qū)τ趦?yōu)化治療方法將是有價(jià)值的,。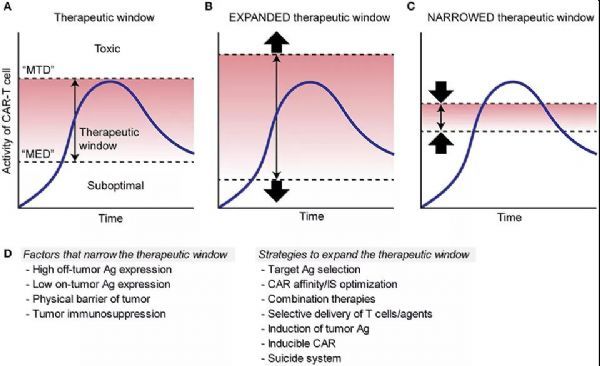
圖解說明CAR-T細(xì)胞療法的治療窗
圖A:小有效劑量(MED)和大耐受劑量(MTD)之間的范圍,;
圖B:在CAR-T細(xì)胞療法中,僅在腫瘤細(xì)胞上表達(dá)的靶向抗原或僅在非關(guān)鍵組織上表達(dá)的抗原擴(kuò)大了治療窗,,因?yàn)椴粫χ匾M織產(chǎn)生直接毒性,;
圖C:靶向在關(guān)鍵正常組織/細(xì)胞中表達(dá)的抗原通過降低MTD使治療窗變窄。
不能僅基于抗原表達(dá)譜來解決治療窗的確定,。例如,,即使在腫瘤和正常組織的抗原表達(dá)差異很大的情況下,其中抗原在腫瘤中以較高密度表達(dá),,但由于內(nèi)在的免疫抑制,,腫瘤可能仍然比正常組織對CAR-T細(xì)胞更具抗性。正常組織中不存在的TME,。在這種情況下,,腫瘤抑制T細(xì)胞浸潤或誘導(dǎo)T細(xì)胞功能減退將通過增加MED來縮小CAR-T細(xì)胞的治療窗。
鑒于真正的腫瘤特異性靶表面抗原尚未被發(fā)現(xiàn),,TAA可能是我們在可預(yù)見的未來的合理目標(biāo),,因此,對于實(shí)體瘤的治療,,制定擴(kuò)大CAR-T細(xì)胞療法治療窗的策略是必須的,。
擴(kuò)大治療窗的可能方法包括:(1)優(yōu)化CAR密度、親和力和感應(yīng),;(2)優(yōu)化免疫突觸形成,;(3)聯(lián)合治療;(4)CAR-T細(xì)胞和治療劑的局部遞送,;(5)誘導(dǎo)靶抗原表達(dá),;(6)其他修飾。
>>>>
優(yōu)化CAR密度,、親和力和感應(yīng)
盡管增加CAR親和力使得能夠識別與靶密度無關(guān)的抗原,但該作用可能引起嚴(yán)重的副作用,,即on-target/off-tumor毒性,,并降低連續(xù)殺傷靶腫瘤的能力。
因此,,重要的是要制定合理的策略,,以確定理想的CAR親和力。
使用輕鏈交換技術(shù)構(gòu)建親和調(diào)節(jié)的scFv是測量CAR的*親和力的可行方法之一。報(bào)道表明,,CAR-T細(xì)胞的親和力高于確定閾值不一定是必需的,,或者更重要的是,通過產(chǎn)生對同一表位具有不同親和力的CAR,,并鑒定這些表位特異性CAR-T細(xì)胞表現(xiàn)出大的細(xì)胞裂解,、增殖和安全潛力的低親和力。
除了改變scFv親和力外,,調(diào)節(jié)表面CAR表達(dá)水平是誘導(dǎo)理想CAR信號傳導(dǎo)的重要因素,。CAR-T細(xì)胞功能受CAR密度和靶抗原密度控制,其中任一個(gè)的低表達(dá)都可導(dǎo)致CAR-T細(xì)胞的功能和靈敏度有限,。
另一方面,,CAR結(jié)構(gòu)和高CAR密度可能導(dǎo)致產(chǎn)生CAR的連續(xù)信號傳導(dǎo)(強(qiáng)直信號),然后通過增加T細(xì)胞分化,、耗竭和活化誘導(dǎo)的細(xì)胞死亡(AICD)而誘導(dǎo)較差的抗腫瘤作用和T細(xì)胞的體內(nèi)植入,。因此,改善CAR密度同時(shí)保持表達(dá)低于強(qiáng)直信號誘導(dǎo)所需的閾值,,可以誘導(dǎo)足夠的抗腫瘤功效并保持對每種靶抗原和CAR構(gòu)建體的安全潛力,。
此外,可以通過腫瘤細(xì)胞上的兩種不同抗原識別組合抗原,。(1)“AND”邏輯門控CAR:僅在同時(shí)識別兩種單獨(dú)的所需抗原時(shí)才能*地激活CAR-T細(xì)胞,。(2)“OR”邏輯門控CAR:CAR-T細(xì)胞通過表達(dá)兩個(gè)CAR或具有串聯(lián)抗原結(jié)合域的單個(gè)CAR,識別兩種不同抗原中的任一種來驅(qū)動*信號傳導(dǎo),。其中,,“AND”邏輯CAR的工程T細(xì)胞能夠?qū)崿F(xiàn)更具特異性和更安全的靶向,“OR”邏輯CAR有潛力克服由于靶抗原丟失而導(dǎo)致的低靶抗原表達(dá)和腫瘤逃逸,。
另一個(gè)有吸引力的平臺是使用銜接分子開發(fā)“通用”CAR,,以克服抗原表達(dá)中的腫瘤異質(zhì)性并使CAR-T細(xì)胞的活化更具條件性。該平臺轉(zhuǎn)化為臨床的一個(gè)潛在問題是銜接分子的免疫原性,。
>>>>
優(yōu)化免疫突觸形成
如上所述,,與組織性良好的TCR IS的靶眼結(jié)構(gòu)相比,CAR IS更加雜亂,,并且其特征在于Lck排列的多焦點(diǎn)模式,、肌動蛋白環(huán)減少和LFA-1擴(kuò)散分布。與TCR IS相比,,CAR IS的顯著能力實(shí)際上是即時(shí)誘導(dǎo)近端信號傳導(dǎo)和細(xì)胞毒性顆粒的快速遞送,,其由MTOC向CAR IS的更快遷移介導(dǎo)。這些優(yōu)勢使CAR-T細(xì)胞能夠快速從破壞的腫瘤細(xì)胞中解離,,并介導(dǎo)有效的連續(xù)殺傷,。
近,,一些研究報(bào)告了關(guān)于CAR設(shè)計(jì)如何影響IS形成的重要發(fā)現(xiàn)。有研究使用由CD28或4-1BB共刺激結(jié)構(gòu)域構(gòu)建的CD19特異性CAR檢測CAR IS質(zhì)量,,并確定CD28加4-1BB的第三代CAR優(yōu)于基于CD28的第二代CAR,,包括IS的結(jié)構(gòu)、信號和功能,。與雙特異性CAR-T細(xì)胞相比,,對兩種神經(jīng)膠質(zhì)瘤相關(guān)抗原HER2和IL13Rα2特異的CAR表現(xiàn)出顯著更高的F-肌動蛋白積累和MTOC極化得增加。
CAR IS的結(jié)構(gòu)特征現(xiàn)在正在闡明當(dāng)中,,其調(diào)節(jié)將是CAR-T細(xì)胞治療的一個(gè)很好的選擇,。已經(jīng)有幾種通過免疫調(diào)節(jié)藥物(IMiDS)改善CAR IS的嘗試,例如來那度胺-沙利度胺的合成衍生物,。來那度胺通過增加肌動蛋白的積累提高了CAR功效,,是一種有潛力增強(qiáng)CAR活性的聯(lián)合療法。
>>>>
聯(lián)合療法
聯(lián)合療法是一種通過克服腫瘤異質(zhì)性和擴(kuò)大治療窗來推動CAR-T細(xì)胞治療實(shí)體瘤的有希望的策略,。
溶瘤病毒(OVs)是用于治療實(shí)體瘤的有希望的藥劑,。OV特異性地靶向腫瘤細(xì)胞,同時(shí)對正常細(xì)胞不起作用,,促使直接的腫瘤溶解,,并激活免疫系統(tǒng)。此外,,還可對OV進(jìn)行基因修飾,,使其在TME中選擇性地表達(dá)治療性轉(zhuǎn)基因。OV在局部表達(dá)治療性轉(zhuǎn)基因的同時(shí)恢復(fù)抗腫瘤免疫反應(yīng)的能力提供了與CAR-T細(xì)胞療法組合的合理理由,。
其他組合方法包括與對4-1BB共刺激受體特異的激動性抗體組合,,其可以直接激活CAR-T細(xì)胞并且還可以減少宿主免疫抑制性免疫細(xì)胞,例如Tregs或MDSC,。
>>>>
CAR-T細(xì)胞和治療劑的局部遞送
CAR-T細(xì)胞的全身性遞送可能會受到對實(shí)體瘤的可及性的限制,,還涉及安全性問題。因此,,直接施用(局部給藥)到腫瘤部位中是CAR-T細(xì)胞遞送的一種解決方法,。另一種方法是設(shè)計(jì)CAR-T細(xì)胞僅在腫瘤部位或主要在腫瘤部位起作用。
為了克服免疫TME,,細(xì)胞因子和趨化因子,,如IL-18、IL-12的局部遞送,,CCL19和IL-7的結(jié)合或表達(dá)檢查點(diǎn)阻斷劑的CAR-T細(xì)胞,,可以幫助克服TME對T細(xì)胞浸潤和功能的阻礙。臨床前模型中已經(jīng)證明了這些方法具有增強(qiáng)的治療功效,,同時(shí)能避免全身不良事件,。
>>>>靶抗原表達(dá)的誘導(dǎo)
正如之前說的,靶抗原密度可以控制CAR-T細(xì)胞療法的功效,。
此外,,靶抗原的丟失或下調(diào)是腫瘤逃逸的主要原因。靶細(xì)胞上抗原表達(dá)的誘導(dǎo)或再誘導(dǎo)可能是擴(kuò)大治療窗的有吸引力的方法,。
據(jù)報(bào)道,,亞致死劑量的輻射可誘導(dǎo)腫瘤細(xì)胞上TAA(如間皮素和CEA)的表達(dá)。此外,,表觀遺傳控制也可以調(diào)節(jié)靶抗原表達(dá),,例如,抗甲基化藥物氮雜胞苷(5-AZA)可以在治療后重新誘導(dǎo)淋巴瘤細(xì)胞上的CD20表達(dá),,包括用靶向CD20的利妥昔單抗治療后,。
>>>>其他修飾
CAR-T細(xì)胞裝備zi殺系統(tǒng),例如誘導(dǎo)型caspase-9 (iCas9)或共表達(dá)截短的EGFR,,將加強(qiáng)CAR-T細(xì)胞的安全性,。這些系統(tǒng)可以通過施用相關(guān)藥劑引發(fā)細(xì)胞固有細(xì)胞凋亡或細(xì)胞外源性抗體介導(dǎo)的細(xì)胞耗竭,從而誘導(dǎo)CAR-T細(xì)胞的消耗,。
可誘導(dǎo)的CAR系統(tǒng),,包括TET誘導(dǎo)系統(tǒng),它能夠使用藥物誘導(dǎo)控制CAR表達(dá),。
合成的Notch系統(tǒng)(synNotch)為CAR-T細(xì)胞的多樣化和靈活性提供了另一個(gè)有吸引力的平臺,。SynNotch受體可以允許在抗原識別時(shí)向T細(xì)胞添加定制應(yīng)答程序。 例如,,synNotch可以在識別抗原時(shí)驅(qū)動定制的細(xì)胞因子分泌,,偏向性的T細(xì)胞分化,或局部遞送治療有效載荷,,如抗體,。
結(jié)語
CAR-T細(xì)胞治療實(shí)體瘤是復(fù)雜且多因素的,具有比用于治療B細(xì)胞白血病和非霍奇金淋巴瘤的CD19靶向更窄的治療窗,。
盡管臨床研究越來越多,,但除了腦室內(nèi)遞送IL13Rα2CAR的膠質(zhì)母細(xì)胞瘤外,很少有明顯的療效,。在這種情況下,,建立擴(kuò)大治療窗的策略顯得至關(guān)重要。
腫瘤生物學(xué),、TME和CAR-T細(xì)胞生物學(xué)仍有許多未知數(shù),。幸運(yùn)的是,解決這些問題的強(qiáng)大工具,,如生物信息學(xué),、質(zhì)譜蛋白質(zhì)組學(xué),、大規(guī)模細(xì)胞計(jì)數(shù)和單細(xì)胞RNA測序等新興技術(shù),將使我們能夠獲得有關(guān)腫瘤,、TME組分和免疫細(xì)胞的信息,。
此外,基因編輯技術(shù)的成熟,,例如CRISPR / Cas9系統(tǒng),,或合成生物學(xué),例如synNotch系統(tǒng),,將能夠幫助靈活地設(shè)計(jì)T細(xì)胞,,有利于CAR-T細(xì)胞治療實(shí)體瘤的突破。
當(dāng)向患者施用新的CAR-T細(xì)胞療法時(shí),,已經(jīng)報(bào)道了幾個(gè)具有意外嚴(yán)重毒性的病例,。不幸的是,目前的技術(shù)不允許我們預(yù)測臨床環(huán)境中的所有毒性,。因此,,目前只有臨床試驗(yàn)可以揭示ACT的安全性和有效性信息。未來,,持續(xù)開發(fā)和改進(jìn)可以預(yù)測毒性的臨床前模型,,以及合理規(guī)劃和實(shí)施臨床試驗(yàn),對于CAR-T細(xì)胞療法的進(jìn)一步發(fā)展至關(guān)重要,。
參考出處:
frontiersin.org/articles/10.3389/fimmu.2018.02486/ful
science.sciencemag.org/content/359/6382/1361/tab-figures-data
(空格分隔,最多3個(gè),單個(gè)標(biāo)簽最多10個(gè)字符)

 12
12






立即詢價(jià)
您提交后,專屬客服將第一時(shí)間為您服務(wù)